Equilibrium Molecular Dynamics (MD)
Molecular dynamics (MD) is a computer simulation method addressing the time development of all participating interacting atoms and molecules according to the laws of Newton and classical physics. Quantum mechanical effects are effectively included via the force fields governing the dynamics but are not made explicit. The method of MD is used in the domains of chemical physics, materials science, and biophysics.
A widespread computational tool that allows for the simulation of molecular dynamics and which is used in the current section is NAMD (NAnoscale Molecular Dynamics). It is a freeware package known for its efficiency in parallel computation that can be used for simulation of comparatively large molecular systems.
- NAMD needs for operation a protein data bank (pdb) file that stores the atomic coordinates and optionally a protein structure file (psf) that stores structural data of the protein such as bond, angle and dihedral information:
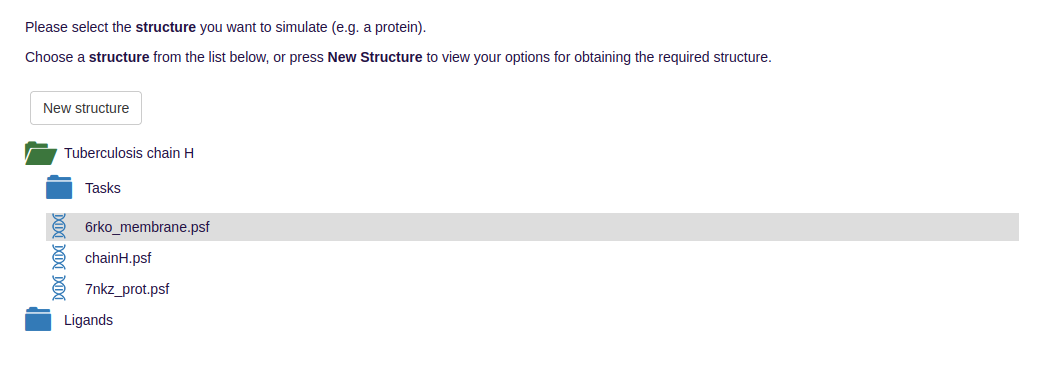
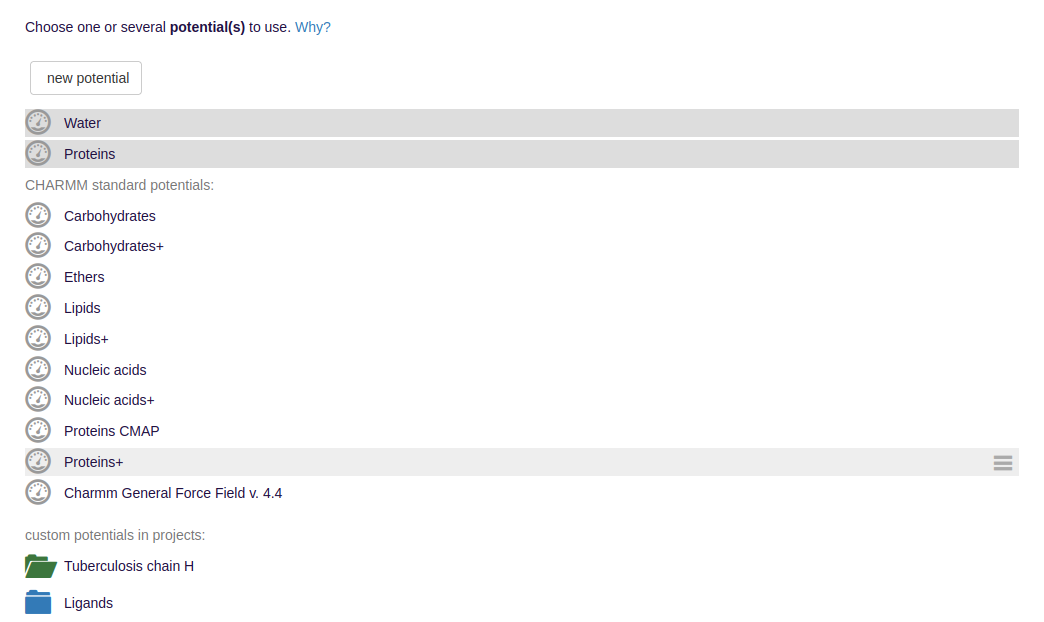
- The potentials are either given by externally uploaded force field parameter files encoding, e.g., for the bond strengths, or are selected from preinstallations.
Among others, typical examples for sets of force fields for molecular dynamics are given by the CHARMM or GROMACS simulation packages.
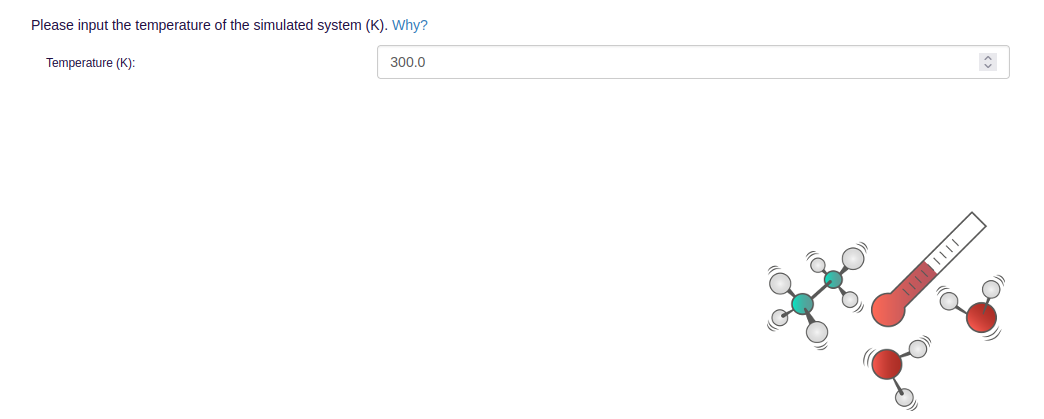
- In order to run MD simulations, general simulation parameters like simulation length, temperature, and pressure need to be specified:

In NAMD, the defined system runs through stages of preparation, energy minimization, various levels of equilibration including increasingly more constituents, and a real simulation part.
The data from the real simulation in the presumed equilibrated state can then be used for various types of further analyses.
- You may further adjust simulation parameters on the task overview page.
Tutorial tasks
Equilibrium Molecular Dynamics (MD) of Villin headpiece in vacuum
Description of the studied system
Villin headpiece is a small fragment of a larger Villin protein. The headpiece consists of a fast and independently folding three-helix bundle that is stabilized by hydrophobic interactions. The headpiece domain is a commonly studied protein in MD due to its small size and fast folding kinetics and short primary sequence.
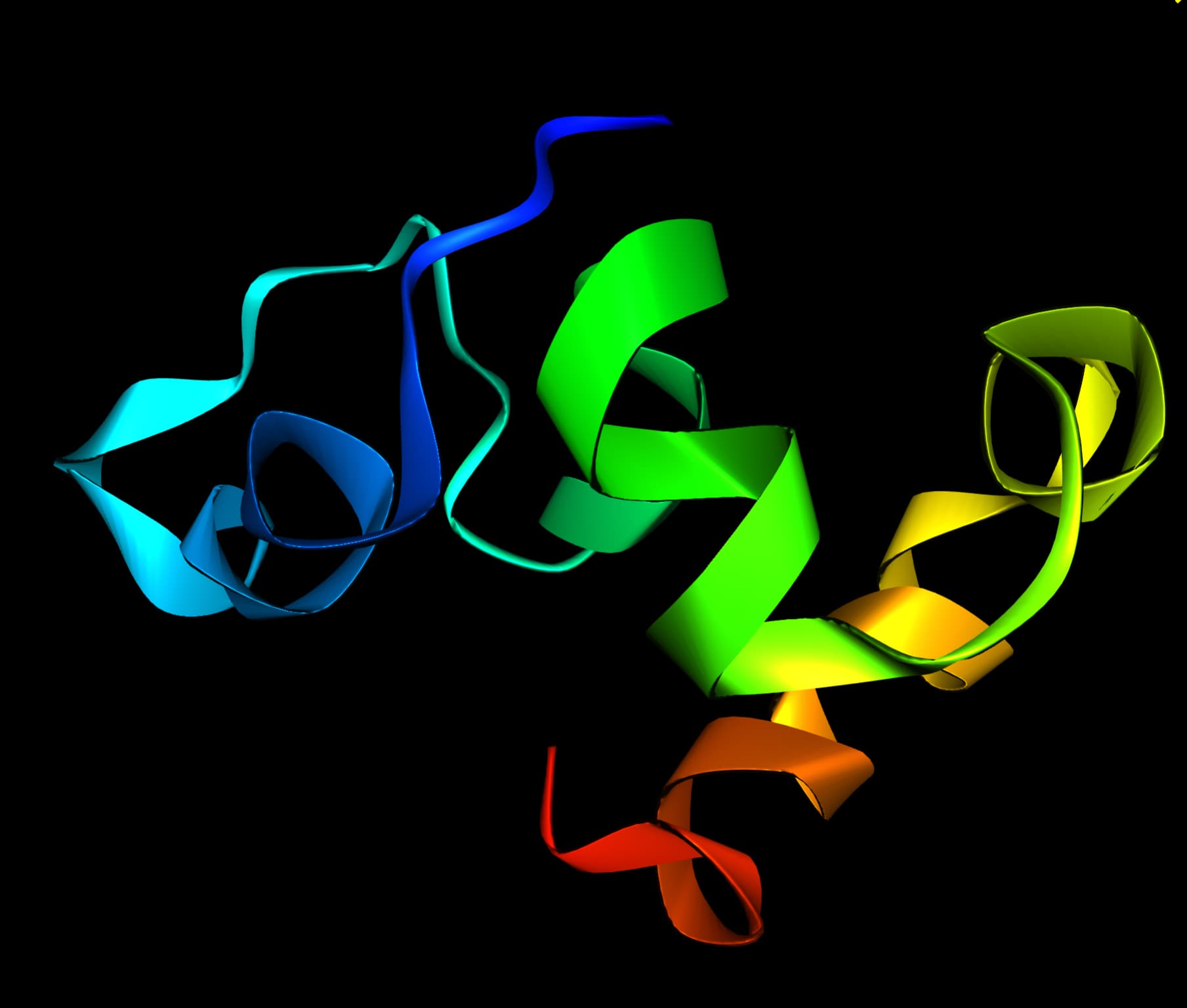
Purpose of the task
For a simple molecular example it can be seen that the time development of the protein fragment can be visualized and it is demonstrated that in principle structural arrangements of proteins can be tracked quantitatively.
Task with files and parameters to be used
Study dynamical evolution of the villin headpiece through MD simulation. Set up an Equilibrium MD simulation of the protein fragment by using the crystal structure provided (villin_headpiece.pdb) and prepare the simulation for a protein in vacuum, i.e. do not solvate the molecule or add salt. Set simulation temperature to 310 K and pressure to 1 bar. Choose a simulation time of 0.1 ns.
The simulation is divided in four stages: Minimization and equilibration 1, Equilibration 2, Equilibration 3, and Production. You will see the corresponding tabs in the VIKING wizard summary page before you start your simulation. In order to obtain some visual results fast (i.e. in <10 mins) you need to modify the standard settings in Stages 1-3: In stage 1 change the number of minimization steps to 100, simulation length to 0.01 ns; In stages 2 and 3 set simulation length to 0.01 ns. Do not change the simulation time in stage 4.
Estimated runtime of this example task
Less than 7 minutes.
Expected results
A cartoon representation of the protein is shown and by scrolling through its time evolution some motion of it in vacuum can be observed in the Viking visualizer. The time evolution of the total energy, kinetic energy, potential energy, and temperature is shown, however, the fluctuations are very large.
Equilibrium Molecular Dynamics (MD) of Trp-cage miniprotein in solution
Description of the studied system
Trp-cage is a synthetic 20-residue miniprotein, which folds rapidly and spontaneously to a well-defined globular structure. Due to its small size and fast folding, it is an ideal model system for experimental and theoretical investigations of protein folding mechanisms.

Purpose of the task
Besides the example of the Villin protein here is another simple example for which the time development of the protein can be visualized and demonstrated that in principle structural arrangements of proteins can be tracked quantitatively. A little folding activity of the protein might be guessed from the time evolution.
Task with files and parameters to be used
Study dynamical evolution of the Trp-cage miniprotein through MD simulation. Set up an Equilibrium MD simulation of the protein (Use the PDB id 1L2Y in the wizard to automatically download the needed structure). Solvate the molecule in water, by defining a solvation box with the padding distance of 7 Å in all directions. Use salt concentration of 0.1 mol/L, set simulation temperature to 310 K and pressure to 1 bar. Choose a simulation time of 0.1 ns.
The simulation is divided in four stages: Minimization and equilibration 1, Equilibration 2, Equilibration 3, and Production. You will see the corresponding tabs in the VIKING wizard summary page before you start your simulation. In order to obtain some visual results fast (i.e. in <10 mins) you need to modify the standard settings in Stages 1-3: In stage 1 change the number of minimization steps to 100, simulation length to 0.01 ns; In stages 2 and 3 set simulation length to 0.01 ns. Do not change the simulation time in stage 4.
Estimated runtime of this example task
Less than 4 minutes.
Expected results
A cartoon representation of the protein in solution is shown and by scrolling through its time evolution some motion of it can be observed in the Viking visualizer. Viewing the system’s components (protein or solvent) can be turned on and off independently allowing to show different representations of the system. In the time evolution the temperature and the kinetic energy plots show the same characteristics.
Literature
R. Zhou, Trp-cage: Folding free energy landscape in explicit water, PNAS, 2003.
Official NAMD tutorials
NAMD tutorial (available at http://www.ks.uiuc.edu/Training/Tutorials/)